搜索

 英国上市公司官网365
英国上市公司官网365

 学院概况
学院概况

 师资队伍
师资队伍

 人才培养
人才培养

 招生就业
招生就业

 科学研究
科学研究

 平台基地
平台基地

 党群工作
党群工作

 校友之家
校友之家

 安全工作
安全工作

 网上办事
网上办事



 当前位置:
当前位置:酿酒酵母是首个基因组被完整测序和注释的单细胞真核模式生物,广泛应用于细胞生理学、合成生物学和系统生物学等研究领域。基因组尺度规模代谢网络模型(genome-scale metabolic network models, GEMs)是全面表征目标菌株生化反应网络知识的基础模型,已成为解析细胞基因型-表型关系的重要计算工具之一。
近日,英国上市公司官网365鲁洪中课题组联合国内外多家单位,整合过去五年的酵母分子生物学和生理学研究,发布了新一代酿酒酵母基因组规模代谢网络模型——Yeast9,为酵母的系统生物学研究和生理学研究等提供了可靠的计算工具。该成果以“Yeast9: a consensus genome-scale metabolic model for S. cerevisiae curated by the community”为题发表在《Molecular Systems Biology》上。上海交通大学与华东理工大学联合培养研究生张承昱为第一作者,英国上市公司官网365鲁洪中副教授和查尔姆斯理工大学Eduard J Kerkhoven博士为该论文的共同通讯作者。
相较于上一代模型(Yeast8),Yeast9的主要改进包括:(1)进行了一系列有针对性的数据扩展,提高了代谢网络的覆盖范围,为模型增添了29个新基因,202个新反应,以及139种新代谢物;(2)通过多轮人工校正,提高了代谢网络模型中基因-蛋白质-反应(gene-protein-reaction, GPR)关系和代谢物注释的准确性,增强了模型的准确性和可靠性;将每个反应分配到明确的代谢途径子系统中,便于可视化分析;(3)根据从多个数据库收集到的数据,为绝大多数代谢物和反应新增了热力学信息,并平衡了模型中大多数质量或电荷不平衡的反应(图1)。
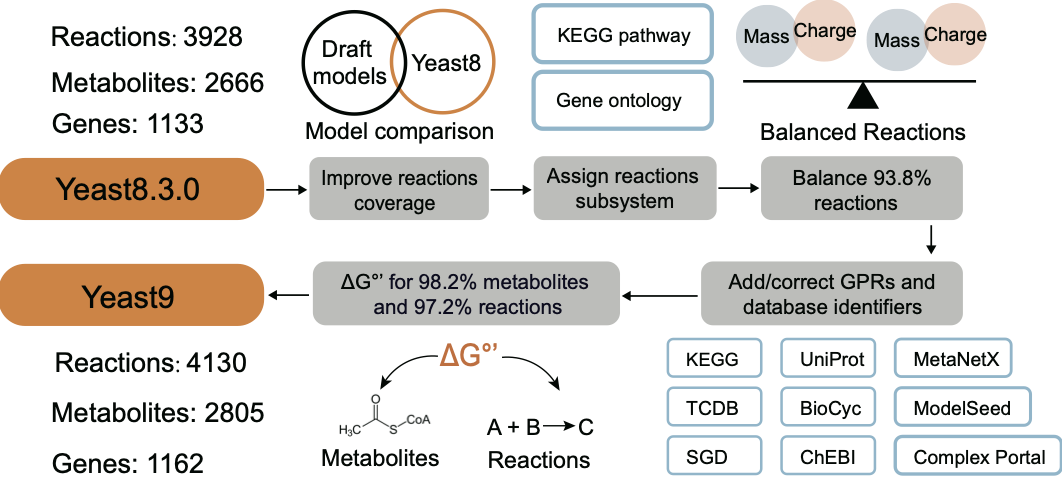
图1.Yeast9相较于Yeast8 的主要改进。Yeast9模型包含1162个基因,2805种代谢物和4130个反应。将Yeast8与使用RAVEN构建的草稿模型相比较,发现新的反应。为几乎所有代谢物和反应添加了ΔG°’。根据来自KEGG或SGD的途径注释,将每个反应与单一子系统关联。通过与数据库进行多轮人工对比,添加或修正了各种GPR。几乎对所有反应进行了校订以确保质量和电荷平衡。
与Yeast8相比,Yeast9在必需基因预测、双基因敲除表型预测和底物利用预测等方面的表现有了一定程度的改进;此外,新增的热力学信息使得探索代谢中物质转化的驱动力成为可能(图2)。为了系统评估Yeast9的预测性能,本研究还进行了以下工作:(1)基于酵母细胞在高渗透压或对照条件下的转录组数据建立163个条件特异性的GEMs,发现利用随机森林和主成分分析等机器学习方法能够区分在高渗透压和正常条件下的单细胞代谢网络,此外,还发现在相同条件下不同细胞的代谢网络存在异质性。(2)使用Yeast9模型计算酵母对不同氮源的偏好分数,通过多组学分析揭示在氮限制条件下酵母代谢重构的情况。(3)利用大规模的转录组数据约束Yeast9,创建了1229个单基因敲除菌株的菌株特异性GEMs(strain-specific GEMs,ssGEMs),通过这些ssGEMs成功预测了不同基因敲除菌株的生长率和敲除基因的功能。作者预计通过转录组、蛋白组等组学数据的整合,Yeast9有望成为一款被广泛使用的主流细胞代谢模型,为酿酒酵母和其他非常规酵母(如毕赤酵母、解脂耶氏酵母)的系统生物学研究和合成生物学应用提供了宝贵的计算资源。
.png)
图2.对Yeast9预测能力的系统性评估。Yeast8与Yeast9在预测基因必需性及Memote评分之间的比较(A)。Yeast8与Yeast9在碳源、氮源、磷源和硫源利用方面的对比 (B)。在有氧和无氧条件下的生长模拟 (C)。Yeast 9能根据“与/或”的关系预测两个基因组合的合成致死性后果,准确率达到80% (D)。忽略“与/或”的关系,Yeast9在预测合成致死性方面的准确率为59% (E)。TCA、EMP和PPP中的ΔG°’概况。颜色表示ΔG°’的值。红线意味着反应在热力学上是可行的;蓝线表明反应在热力学上是不可行的。粗边圆角矩形内的数字代表从葡萄糖合成乙酰辅酶A、丙酮酸、谷氨酰胺、天冬氨酸和乙醇时TCA、EMP、PPP及相关反应途径的总ΔG°’(F)。
值得注意的是目前大部分GEMs因缺乏蛋白质合成、酶丰度和酶动力学的约束,难以准确预测基因操作对细胞生长和目标产物合成的定量影响,限制了更高效的细胞工厂设计算法开发。为此,人们提出了包括动力学模型、酶约束模型、多尺度模型和全细胞模型在内的更先进的代谢模型,以增加代谢模型的应用场景。这些更先进的模型的成功构建需要一定数量的参数,例如酶的转换数(kcat)。然而,测定细胞中数以千计的酶的kcat值是一个艰巨的任务。针对该问题,鲁洪中课题组结合前沿深度学习算法,开发了新的预测酶kcat值的工具DeepEnzyme。具体而言,DeepEnzyme利用最新的深度学习算法Transformer和图卷积网络,提取来自底物、蛋白质一级序列和蛋白质三维结构的特征信息,进而实现酶kcat高通量预测与分析(图3)。
.png)
图3.DeepEnzyme用于预测kcat的框架。DeepEnzyme整合了变换器和图卷积网络(Graph Convolutional Network,GCN)模型,从酶和底物中提取特征以预测kcat值。GCN用于根据蛋白质三维结构和底物邻接矩阵提取结构特征;变换器则用于从蛋白质序列中提取序列特征。
相较于以往的预测酶kcat的深度学习模型,DeepEnzyme在处理与训练数据集中酶序列相似度较低的序列信息时表现出显著的鲁棒性,证明蛋白质3D结构特征信息可以有效提升酶kcat预测的精准性和稳健性(图4)。此外,DeepEnzyme还能够识别影响催化功能的关键残基位点。总之,DeepEnzyme的开发将有助于在蛋白组规模解析酶的动力学参数,从而加速各类先进细胞数字模型构建,同时有望促进对不同物种中酶的功能及其进化模式的理解,推动理性蛋白质工程的发展。
.png)
图4.DeepEnzyme 在kcat预测中的性能相比现有模型有所提升,即使对于测试数据集中的蛋白质序列与训练数据集中的序列相似度较低的情况也是如此。不同模型在测试数据集上的R2值比较 (a)。不同模型在测试数据集上的RMSE值比较 (b)。DeepEnzyme、TurNuP、DLkcat和DLTkcat在测试数据集中不同序列相似度水平下对酶的kcat值预测的R2比较 (c)。来自黏质沙雷氏菌和枯草芽孢杆菌的两种酶,它们的EC编号均为1.3.3.4,在蛋白质三维结构上高度相似(TM score = 0.8762),其中黏质沙雷氏菌的酶用灰色表示,枯草芽孢杆菌的酶用红色表示 (d)。上述两种酶的氨基酸序列相似度为27%(Q表示来自黏质沙雷氏菌的酶,T表示来自枯草芽孢杆菌的酶) (e)。
该项工作以“DeepEnzyme: a robust deep learning model for improved enzyme turnover number prediction by utilizing features of protein 3D-structures”为题发表《Briefing in Bioinformatics》上。上海交通大学与重庆理工大学联合培养硕士王彤为第一作者;英国上市公司官网365鲁洪中副教授为论文的通讯作者,英国上市公司官网365为第一和责任通讯单位。
鲁洪中课题组长期从事于高精度数字细胞模型构建,破译基因型-表型关系;开发细胞工厂精准设计算法,实现BT和IT技术的深度结合,以期解决细胞代谢全局可预测、菌株理性改造等合成生物学领域的关键问题与挑战。相关成果以一作或通讯作者发表于Nat. Commun.、Mol. Syst. Biol.、Brief. Bioinform.、Metab. Eng.和Trends. Biotechnol.等国际知名期刊。基于鲁洪中博士的研究成果,共有6项科研成果专利正在申请中。以上两项研究获得国家重点研发计划(2022YFA0913000)、上海市浦江人才计划以及国家自然科学基金(22208211和22378263)等项目的资助。
相关论文链接:
《Molecular Systems Biology》:https://doi.org/10.1038/s44320-024-00060-7
《Briefing in Bioinformatics》:https://doi.org/10.1093/bib/bbae409

版权所有 英国上市公司官网365-英国正版365官方网站